点击微信咨询
1364994806  (扫码)
(扫码)
私人打胎药商城在线购买,回头率最高/全国都包邮/货到付款正品/
时代发展,但是很多不和谐的音符也应运而生,孩子们越来越早熟,据调查很多医院打胎的年龄越来越趋于低龄化,还有些女性意外怀孕之后,因为没有结婚年龄小不敢和家里说,自己吃打胎药打胎,哪种情况可以吃打胎药进行打胎,吃打胎药后会出现哪些反应?但是药物流产不是适用所有人,还是要到医院进行检查,不然会出现生命危险。 下面,一起来详细了解下关于吃打胎药的相关问题吧。
hY112.ZHilI123。cOm
私人打胎药商城在线购买 ,添加文内微信免费咨询!
郑重承诺:堕胎药、堕胎药米非司酮片全国范围顺丰速递保密派送,全程指导用药!(详情请咨询)
专业药师在线指导,合理用药,更健康,私人打胎药商城在线购买 ,在线咨询,承诺正品全国包邮。
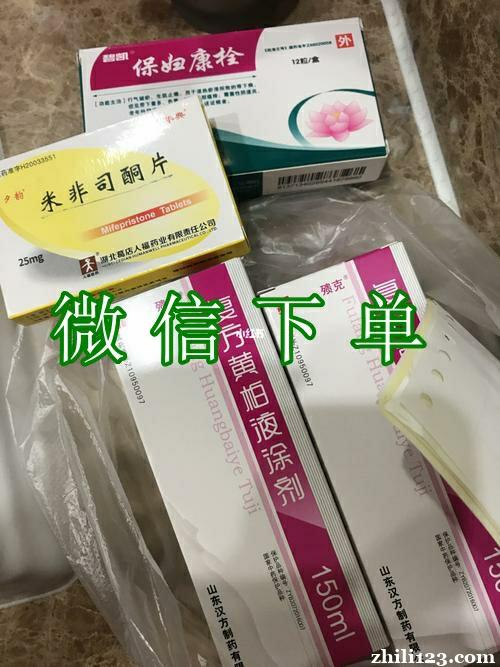
私人打胎药商城在线购买 -药品介绍
商品名称 米非司酮
通用名称 米非司酮
英文名 Mifepristone
批准文号 国药准字H
规格 ----
用法用量 口服给药:
1.终止早孕:停经49天内的健康早孕妇女,服用米非司酮方案有两种:
(1)顿服200mg;
hY112.ZHilI123。cOm
(2)每次25mg,每天2次,连续3天。第3或第4天清晨于阴道后穹隆放置卡前列甲酯栓1mg(1枚),或使用其他同类前列腺素药物,卧床休息1h后再起床,以免药物流出。如使用米索前列醇口服片,则服用400~600μg(2~3片),在门诊观察6h,注意用药后出血情况,有无胎囊排出和不良反应,并且必须按药物流产常规的要求进行观察和随访。
2.用于中、晚期胎死宫内:每次200mg,每天2次,或每次600mg,每天1次,连服2天。
分类 化学药品
剂型 原料药
外用药 否
国家/地区 国产
生产企业 湖北葛店人福药业有限责任公司
私人打胎药商城在线购买 -不良反应
部分早孕妇女服药后有轻度恶心、呕吐、眩晕、乏力和下腹痛。极个别妇女可出现潮红、发热及手掌瘙痒,甚至过敏性休克。
私人打胎药商城在线购买 -药理
hY112.ZHilI123。cOm
米非司酮为受体水平抗孕激素药,具有终止早孕、抗着床、诱导月经及促进宫颈成熟等作用,与孕酮竞争受体而达到拮抗孕酮的作用,与糖皮质激素受体亦有一定结合力。米非司酮能明显增高妊娠子宫对前列腺素的敏感性。小剂量米非司酮序贯合并前列腺素类药物,可得到满意的终止早孕效果。
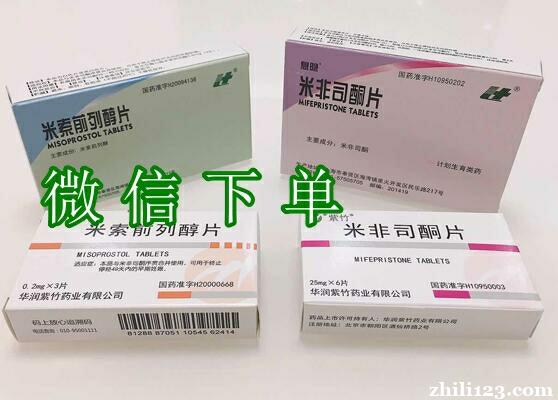
私人打胎药商城在线购买,回头率最高/全国都包邮/货到付款正品/
如果怀孕了,确实会出现一系列的征兆,比如停经,出现早孕反应,尿频,乳房发生变化等。
1.停经。如果平时月经周期比较规律,有性生活的女性如果出现月经推迟的情况,就需要考虑是否怀孕。如果月经推迟时间在10天以上,要高度怀疑怀孕。
2.早孕反应。一般在停经6周左右的时候,女性可能会出现一些不适症状,比如畏寒怕冷、头晕、身体乏力、嗜睡、恶心、呕吐、食欲不振等,有的患者可能会出现情绪改变。
3.尿频。在怀孕以后,子宫就会逐渐增大,就有可能会压迫到膀胱,导致出现尿频症状。
4.乳房变化。怀孕后雌激素和孕激素水平升高,会导致出现乳房胀痛症状,而且乳房体积逐渐增大,乳头增大,乳晕颜色加深,乳晕周围出现蒙氏结节。如果是哺乳期女性怀孕,乳汁会明显减少。
人流后要吃米非司酮片,这个药有用吗?
问题描述:前段时间陪朋友去人流,之后医生开了米非司酮片,是不是用来终止妊娠的?
问题分析:
米非司酮为受体水平抗孕激素药,具有终止早孕、抗着床、诱导月经及促进宫颈成熟等作用,与孕酮竞争受体而达到拮抗孕酮的作用,与糖皮质激素受体亦有一定结合力。米非司酮能明显增高妊娠子宫对前列腺素的敏感性。小剂量米非司酮序贯合并前列腺素类药物,可得到满意的终止早孕效果。一般人流术后如果宫内残留超过1.0.药物治疗效果不太好,大多需要进行清宫治疗,小的残留可在医生指导下适量服用药物,这期间建议您注意休息,适当加强营养。这个是激素类的,具有终止妊娠和调理女性内的雌激素,术后吃的话,是调理激素的药。吃这个是没事的,短时期内没有什么副作用。
意见建议:
一定要做好安全措施,不能麻痹大意或者怀有侥幸心理。空腹或进食2小时后口服1片,服药后禁食1-2小时,也可以在医生的指导下服用。
私人打胎药商城在线购买,回头率最高/全国都包邮/货到付款正品/
私自药流的危害
私自药流的危害巨大,虽然有一小部分人自己通过药流成功地完成了流产过程,并且身体没有什么大的危害,但是这种行为是绝对要禁止的。药流的人千万不要抱有侥幸心理,一定要按照正规的方式进行操作。首先药流适用于停经49天以内或者是正常怀孕5到7周的女性。其次,药流之前必须有B超检查以便于确定是否有宫外孕,宫外孕不能做药流。最后,私自做药流一旦出现了大量失血或者是严重感染的情况,自己根本就没有办法处理,很有可能危及生命。
1、 所以药流千万不要私自做,一旦出现了危急情况后果是难以估量的。并且私自做药流很难自己判断出是否流干净,如果没有流干净并且没有做相应的处理,那么除了对身体有危害之外,对于下次妊娠也是有极其重大的影响,甚至还有可能导致各种意想不到的妇科疾病。
2、 正确的药流方式是停经49天以内,并且通过B超检查确诊为已经怀孕,没有出现宫外孕的现象,并且患者的身体没有肝脏以及肾脏等方面的重大疾病,然后去医疗设施具备的医院在医生的指导下进行药流。
hY112.ZHilI123。cOm
3、 药流服用药品之后2到3天时间胚囊就会通过阴道排出体外,并且在此期间出血的量会比较大,最好此段时间之类有医生的观察指导,以便于在出现危险的情况下有应对的措施。通常服用药品之后两周左右时间蜕膜组织才会排出体外,阴道流血的现象才会停止。
注意事项:
私自服用药品做药流是绝对不可取的,因为一旦出现危险不能够及时联系到医院以及医生,有时候即便是联系到医生也会由于不具备各种治疗条件而导致悲剧。
-----------------以下为新闻资讯,可略过!-----------------------
李宁教授:如何破局我国临床研究面临的机遇和挑战?
·中国临床试验实施质量基本达到国际水准,国内核查标准比FDA更严格;
·监管方对科学性的重视提升,是临床试验行业的重大利好;
·新药研发要打破“交棒模式”,全链条融合提高转化率;
·建立专职临床试验团队和研究型病房,重视CRC群体、改善其工作条件。
Article List
近期,中国医学科学院肿瘤医院GCP中心李宁教授等在《中华肿瘤杂志》发表《基于科学监管体系的中国药物临床试验实施质量国际评估比较》一文,对国内临床试验质量的实际情况进行全方位分析探讨。
当前中国临床试验行业现状和质量标准、科学监管和转化创新能力,究竟处于怎样的水平、还有哪些差距和不足?对此,在由中国医药创新促进会指导、艾美达医药咨询承办的中国医药创新政策论坛现场,中国医学科学院肿瘤医院副院长、临床研究国家级质量评价和促进中心(肿瘤领域)常务副主任李宁教授接受了主办方采访。
李宁教授指出,经过多年的实践与努力,中国临床试验科学监管体系、实施质量和效率、优化流程机制等方面已有了很大提升,在一定程度上获得了行业乃至国际认可。但我国临床试验能力仍有不少的提高空间,同时利用新模式提高转化成功率、增强行业教育及监管工作,提升临床试验良好环境,有助于我国临床试验行业的发展,更对研究成果转化具有重要推动作用。
01
中国临床试验的监管变迁
Q:前段时间,关于“中国临床试验质量究竟如何”的话题引发了行业讨论。对此您的看法是?
A:国家药监局在2015年开启的“722核查”,不仅推动了临床试验监管体系的改革,也重塑了整个临床研究行业的角色分配,提高了各方团队对于临床试验实施质量的重视程度。2017年我国加入ICH之后,更是在监管体系上整体对标FDA、EMA和PMDA等国际标准要求,全方位提升监管水平。
随后,在一系列密集出台的法规和指导文件的引导下,我国也进一步建立起一套具有中国特色的临床试验实施质量监管体系。CFDI核查不仅关注项目和申办方,也将临床试验机构和研究者作为重点对象,且设有中国特有的机构日常监督检查,充分利用机构负责制和主要研究者(PI)负责制双保险机制的优势,强化对临床试验全流程的监督监管。另外,在核查队伍的组成方面,我国核查专家通常情况下会有4-5人,包括一位核查组长,通常来自CFDI,组员则可能来自于医院GCP办公室或是相关领域人员,同时为了公正公平,省/市级的药监部门通常会委派观察员观摩整个核查过程。
在监管环境得到明显优化的情况下,我国临研行业各方团队也在积极学习制度法规,提高自身规范性,尽可能保证临床试验实施过程的质量及试验数据的真实可靠。
Q:近期,您牵头发表的《基于科学监管体系的中国药物临床试验实施质量国际评估比较》一文,对国内临床试验质量也进行了全方位分析探讨。请您谈一下我国临床试验实际上处于什么水平?
A:评价临床试验质量不能空口无凭,我们可以通过FDA的核查结果来客观的看待国内的临床试验质量。
FDA现场核查的结论分为“不需要采取行政措施”(NAI)、“发现问题,自愿采取行政措施”(VAI)和“严重问题,需要采取官方行政措施”(OAI)三类。根据我们整理的数据来看,2009年至2023年7月,我国机构共接受FDA核查45次,2016年之后相较前期实施质量逐年提升的趋势明显,结果均为合格,严重问题零发生,NAI由48.0%提高至85.0%,逐渐高于FDA核查美国本土机构的同期结果(71.1%),仅有的两次OAI问题均发生于2016年之前。
同比美国核查本土的数据可以看出,如果说“722”之前中国的临床试验实施质量与国际水平尚有一定差距,但近年来在行业各方的努力之下已经有了明显的提升,逐渐达到和符合国际水平和标准。而且FDA进行核查的医院并不均是像我院这样的国家级医学中心,他们更多核查的是省级,甚至是省级以下的肿瘤医院或者综合医院,这说明我们国内临床试验质量的提升不是偶然的、个别的,而是整体的,平均的。
Q:目前中国的临床试验数据受行业认可程度如何?
A:中国临床试验数据认可度可以从我国参研的上市新药中得到验证。2016年后,越来越多的中国临床试验数据及结果为新药获批上市提供理论支撑,利用中国临床试验数据支持FDA和NMPA批准上市的抗肿瘤新药数量也逐年增多。
以抗肿瘤新药为例,过去10年中我国有100余种抗肿瘤新药成功上市,经过临床应用和市场的检验,至今尚无撤市,中国药企开发的抗肿瘤新药被纳入专业指南的次数也持续增加。中国药企的37种新药(58.7%)被纳入2022版CSCO指南,15种新药(23.8%)被2023版NCCN指南收录。可以看出,中国的临床试验数据质量在临床实践使用中逐渐被业界所接受。
我自己在实际工作中也有比较深刻的体会。在与其他国际医疗机构、企业交流的过程中,我们有了更多的话语权,更多的企业认可我国的临床研究数据,将更多重要的临床试验项目安排在国内进行,这都是我国临床研究水平提高的力证。
Q:您觉得我们自身还有哪些不足,下一步应向哪些方面加强和发展?
目前中国的临床试验还在快速发展阶段,仍有一些问题需要我们持续关注。例如,尝试利用已经获得的临床试验数据质量结果和质量关键要素,建立基于风险的科学化监管策略,探索临床试验风险预警和分级处理,来补充临床试验全链条的风险管控。我们还应持续推进临床研究能力建设,提升申办方和研究者的研发设计能力,以及研究团队参与人员的科学研究水平,从根本上提高我国临床试验的质量。另外,加强与国际监管和行业的宣传交流合作,提高中国临床试验的行业声誉和国际影响力也十分必要。
02
加强科学监管
是行业重大利好
Q:资本寒冬的当下,市场越来越渴望提高临床转化效率,我国当前的转化效率如何,还需要哪些提升?
A:临床试验最终目标是让更加安全有效的新药上市,为有需要的患者提供更多治疗选择。我们对于产品质量的要求不断提升,相应的研发成本同样也会上升。临床转化并不一定以实体产物的形式出现,可能是一种治疗方案,也可能是一个实体的药品或者器械。但临床转化的逻辑是共通的,就是转化过程中必须有自己的理论被证实,同时也能证伪。
怎样提高这种转化的成功率?首先要认清一个事实,就是临床转化本身就是伴随风险的,而且风险很大。比如CNS三大期刊,每年关于肿瘤相关的论文有5000多篇,但相关的临床试验只有300多项进行。全球每年获批的肿瘤相关适应症50多个,新增药物更是不到20个。
当前,药物研发尚处于一种“交棒模式”中,即前期的科研人员做好了理论,随后就把这个研发工作交接给了下一波工作人员,这样依次交接下去,最后再交到临床试验人员手中。这种模式的弊端在于,每个工种只负责自己的工作,与上下游之间其他工作人员不能形成交流,自然也不会对其他的工作负责。
我们希望能把药物研发工作的链条整合起来,形成类似“多学科诊疗(MDT)”的模式。在药物最初研发设计时,就应该想好最终的目的是什么,也就是要有对应的临床需求,同时结合上下游的工作人员,甚至可以涉及监管层面的人员,共同参与开发策略设计。我相信这种全链条整合式的模式,应该能大大提高我们当前的临床转化率。
Q:临床试验持续创新发展,目前监管的趋势和关注有哪些变化,对临床试验及整个医疗行业来说意味着什么?
A:近期相关部门动作不断。7月初,国家药品监督管理局发布《药物临床试验机构监督检查办法(试行)(征求意见稿)》,对药物临床试验机构日常监督检查工作规范化、制度化。随后,国家药品监督管理局食品药品审核查验中心(CFDI)发布《药物临床试验机构监督检查要点和判定原则(征求意见稿)》,进一步明确相关监督检查工作的要点、细则。7月底,国家药品监督管理局药品审评中心连发三项指导原则,《以患者为中心的药物临床试验设计技术指导原则(试行)》《以患者为中心的药物临床试验实施技术指导原则(试行)》《以患者为中心的药物获益-风险评估技术指导原则(试行)》,围绕“以患者为中心”对临床试验设计、实施及评价全流程进行规范。
目前来看,我们的监管正在逐渐加强科学属性。临床研究本身就是一个专业的学科,相应的监管也要从管理方法、评价标准、监管责任、监管体系等方面适应这种科学属性。当前临床研究行业存在着进步空间。比如,如何判断一个药物的好或者坏,像肿瘤药,是延长PFS(无进展生存期,Progression-free Survival)好,还是延长OS(总生存期,Overall Survival)好?这是一个科学性的问题,需要我们整个行业来探讨、来调整。现在监管已经开始发现这些问题,并制定了相关内容,这对临床研究行业是重大利好。
hY112.ZHilI123。cOm
03
提升临床试验质量与效率
医肿是如何做的?
Q:随着中国临床研究飞速发展和创新能力持续增强,在临床试验开展的不同时期会产生新的机遇和挑战,您作为医疗机构的管理者,遇到了哪些难点问题,对此都尝试了哪些措施?
A:一项临床试验在研究中心开展的过程中,需要经过很多次审查、沟通、协调。所以,程序多是首先被大家诟病的问题,尤其是大医院里科室多、部门细,进了医院大门甚至不知道找谁。所以,我们中国医学科学院肿瘤医院的办法是实行中心化建制,即GCP中心负责所有临床研究工作,提供一站式服务,不需要再跑其他的部门。
对于整个办事流程,我们推出了详细的办事手册,每两年就更新一次,并且列出明确的时间进度表。另外,我们也是国内首家做到接受中心伦理的医院,通过前置立项、前置伦理、合同并行的措施,不断加快临床试验启动前的沟通效率、压缩整体试验的进行周期和成本。
Q:众所周知,医肿作为行业中临床试验承载量非常大的一家研究机构,是如何解决医生常规临床工作和参与临床试验的时间和精力问题的?
A:我们的办法是成立了一支高水平的专职临床试验团队,包括专职研究医师、研究药师、护理以及数统人员,他们是来自国内一流院校、全职投入临床研究的多学科人才。为我院研究者提供全面的临床试验支持和服务,让研究者能将宝贵精力投入到最重要的临床试验决策中,专心做好最关键的受试者的临床治疗判断,重点关注和掌握临床试验中新药的安全性和有效性。
还有硬件设施方面。我们设置了研究型病房,患者可以在这里完成采血、静脉用药、淋巴单采、膀胱灌注等,也不需要跟门诊的患者“抢”床位。目前我们的研究型病房每年收治患者8000多人次,也是全院最繁忙的部门之一。面向受试患者,尽可能的减免程序,可以做到零押金无垫付进行住院,解决患者治疗中的经济困难。
另外,我要特别提一下CRC这个群体,他们是近年来临床试验中非常重要的角色。我们曾经做过一个调查,主要是为了探究试验整体用时以及工时分配,最终结果显示临床试验项目有60%的工作时间需要CRC的支持和协助。为了方便他们工作,我们医院也对CRC进行统一的管理和安排,实行员工化待遇、专业能力培养、配备办公工作条件,今年新增安排了700平米办公场所。
Q:综上,临床试验行业在近几年的改善可谓巨大,那么如今如何让临研行业甚至整个社会逐渐转变对于临床试验的客观认知?
A:临床研究是以患者为中心而展开的研究,这决定了从业者必须跟上患者不断变化的需求,处于长期学习的状态。我们医院针对行业特性开展了许多培训活动,比如面向研究者的培训和面向监管人员的交流。从行业内部提升对临床试验的认知。
其中,我们面向全国研究型医院中突出的青年研究者,提供进阶式的临床研究技能培训。这是一个着眼于5年、10年后的中国创新临床研究,实行小班教学,培训时长至少1年以上。我们希望通过这个活动,将这批青年研究者培养成为未来的Leading PI,为中国的临床研究留下一批人才。
另外,不得不谈的是,受试者招募难一直是临床试验中的困境,公众对于临床试验的印象一直是偏负面的。比如一提到细胞治疗,大家马上就联想到“魏则西事件”。中国的临床试验不仅要得到专业上的认可,同时也需要整个社会大众的认可。我们希望能扭转这种偏见,为此我们在国际临床试验日(5月20日)这天举办公益宣传活动,把临床试验的目的、流程以及能带给受试者的益处讲清楚——参加临床试验,是参与人类医学的发展,能为治疗更多的患者作出贡献。目前,我们已经成立了临床研究方面的患者咨询委员会,围绕着以患者为中心,把患者的真实感受融入临床研究工作中。当我脱下白大褂与患者交流时,身份的变化也让我从更多的角度来理解患者。